I focused mainly on products I might use in my own lab, such as PCR reagents and basic lab products. And there was one company that had something that really made me stop and say “wow!”. ----------- Suzanne
That company was Finnzymes, and the “wow” came from three products that are individually impressive but together make a superb PCR ensemble that is compact, versatile and offers excellent speed and accuracy.
A Compact Thermal Cycler…
First up was the Piko™ Thermal Cycler, which is the smallest thermal cycler. Amazingly, this tiny instrument was around the size of a vortex mixer; with a footprint of only 16cm wide by 17 cm deep and 23 cm high and a weight of only 4 kg. Check out the picture on the right for a comparison of footprint sizes for the Piko™ and a conventional thermal cycler.

Even better, the Piko™ uses a 1/4 of the power of a normal thermal cycler, so this is one way for our experiments to be a bit greener. And it offers signficantly reduced reaction times. More on that later.
The instrument ranges in price from $4500-$6000 depending on the blocks you purchase (there are several options), so it also costs significantly less than traditional thermal cyclers.
A thermal cycler that won’t hog your bench space ticks one box. But your PCR is only as good as your polymerase, and Finnzymes have thought of that too..
A Speedy, Accurate Polymerase…
David Unger, Managing Director of Finnzymes USA, explained that the power behind Phusion™ comes from the unique dsDNA binding domain fused to a Pyrococcus-like proofreading polymerase, which results in a very tight association of the enzyme with the template DNA.
“Taq polymerases work by moving on and off the template. This slows down the enzyme and leads to difficulty if inhibitors are present.
With Phusion™, the enzyme stays linked to the template and so has a faster processivity along with the ability to work in inhibiting conditions such as 20% whole blood in the PCR reaction.”
The enzyme is twice as fast as Taq (<15 second/kb extension time) and can amplify long templates, up to 20 kb with less enzyme. Just as importantly, the error rate is 50 fold lower than Taq and 6 fold lower than standard Pyrococcus furiosus DNA Polymerases.
Sounds great (and it is). Together, Piko™ and Phusion™ offer fast, accurate and economical PCR.
But throw in Finnzymes’ patent pending UTW™ (Ultra Thin-wall) tubes and plates and the whole thing gets a lot more powerful.
96 x 5ul Reactions in 10 minutes…
The UTW™ PCR plates are, as the name suggests, ultra thin, which dramatically cuts down on the time taken to heat and cool the samples.
Piko™ is available with with a 24-well block for UTW™ 0.2 ml tubes, but for maximum output the instrument can be equipped to take Finnzyme’s 96-well UTW™ Piko™ PCR Plates.
These plates are about the size of a microscope slide and Piko™ can take up to 4 of them to make the equivalent of a 384-well plate for easy up and downstream processing with other lab equipment.
Using Phusion™, your 384 x 5 ul reactions can be completed in just 10 minutes flat. Now that’s bench-scale high throughput.
At that speed and size, even the smallest lab can forget about PCR backlogs or downtime.
Courtesy: Suzanne, BitesizeBio











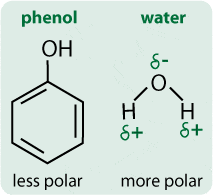 A solvent is a substance, normally a liquid, that can dissolve other substances. Broadly, solvents can be classified according to their polarity, which depends on how extreme the spread of the electron density in the molecule is.
A solvent is a substance, normally a liquid, that can dissolve other substances. Broadly, solvents can be classified according to their polarity, which depends on how extreme the spread of the electron density in the molecule is. The solubility of the proteins is flipped by phenol
The solubility of the proteins is flipped by phenol